图解 AlphaFold
一篇 AlphaFold3 架构的可视化导览,细节和图示可能比你原本想看的还要多。
简介
适合谁读
你想真正弄清楚 AlphaFold3 是怎么工作的吗?它的架构相当复杂,论文里的描述也很密集,所以我们做了这篇更友好、但同样尽量详尽的可视化导览。
本文主要写给机器学习背景的读者,许多地方默认你熟悉 attention 的基本步骤。如果你有些生疏,可以先看 Jay Alammar 的 图解 Transformer。那篇文章是少数能把模型架构讲到单个矩阵运算层面的优秀解释之一,也是本文图示风格和命名方式的灵感来源。
关于蛋白质结构预测的动机、CASP 竞争、模型失败模式、评估争论、对生物技术的影响等,已经有很多很好的解释,所以我们不关注其中的任何一个。相反,我们探索 how(如何).
这些分子在模型中是如何表示的?又经过哪些操作被转换成预测结构?
这篇文章可能比大多数人需要的更详尽;但如果你想看清所有细节,又喜欢通过图示学习,它应该会有帮助 :)
架构概述
首先要注意,AF3 的目标与之前的 AlphaFold 模型有所不同:AF2 主要预测单条蛋白质序列的结构,AF-Multimer 预测蛋白质复合物,而 AF3 可以从序列出发,预测蛋白质以及它可选地与其他蛋白质、核酸或小分子形成复合物后的结构。因此,旧版 AF 模型主要只需要表示标准氨基酸序列,AF3 则必须表示更复杂的输入类型,也就需要更复杂的特征化和 Token 化方案。Token 化会在单独章节中解释;现在只需知道,本文说的 “token” 可能表示一个氨基酸(蛋白质)、一个核苷酸(DNA/RNA),也可能表示一个不属于标准氨基酸/核苷酸的单个原子。
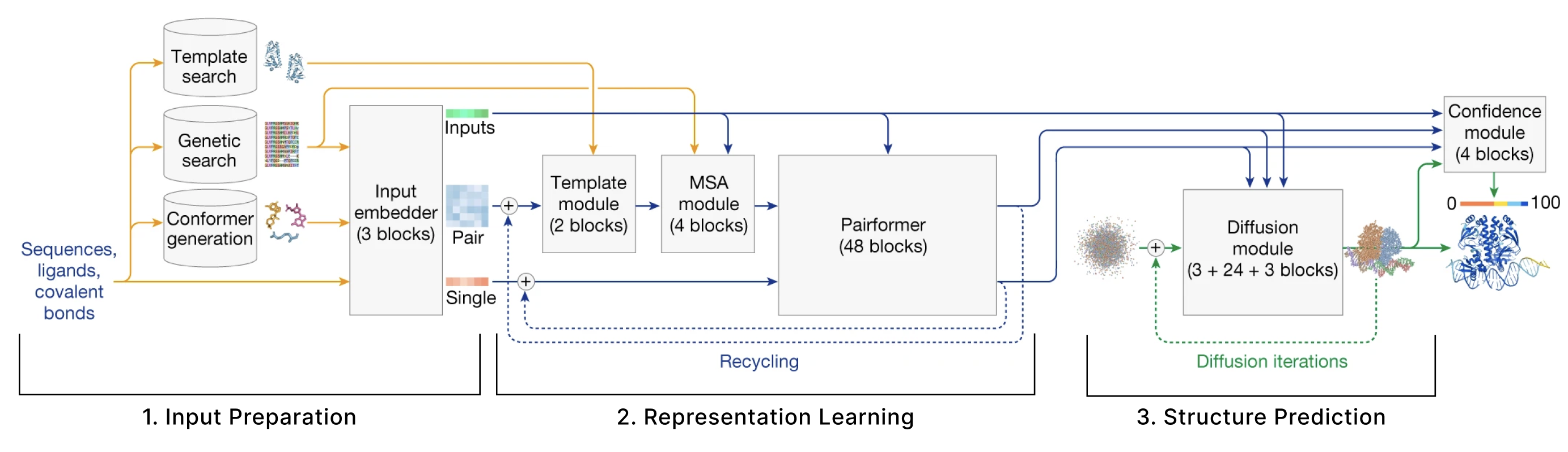
该模型可以分为 3 个主要部分:
- 输入准备 用户提供一些分子的序列来预测其结构,并且这些序列需要嵌入到数值张量中。此外,该模型还会检索其他分子的集合,这些分子被认为与用户提供的分子具有相似的结构。输入准备步骤识别这些分子并将它们嵌入作为它们自己的张量。
- 表征学习 给定第 1 节创建的 Single 张量和 Pair 张量,我们使用许多attention 变体来更新这些表示。
- 结构预测 我们使用这些改进的表示以及第 1 节中创建的原始输入来用条件扩散预测结构。
另外,本文还会介绍第 4 部分:损失函数、置信度和其他相关训练细节 以及第 5 部分: 从机器学习趋势的角度对该模型的一些思考.
变量和图示说明
在整个模型中,蛋白质复合物以两种主要形式表示:“单个”表示,表示蛋白质复合物中的所有 token;“对”表示,表示复合物中所有氨基酸/原子对之间的关系(例如距离、潜在的相互作用)。二者都可以在原子级或 token 级表示,并且将始终以 AF3 论文中的这些名称和颜色显示:

- 这些图示抽象了模型权重,仅可视化激活形状如何变化
- 激活张量总是标有论文中使用的维度名称,并且当这些维度增长/收缩时,图示的大小模糊地遵循。
隐藏维度名通常以 “c” 开头,表示 channel。作为参考,使用的主要尺寸为 cz=128,cm=64,catom=128,catompair=16,ctoken=768,cs=384. - 只要有可能,此(以及每个)图中张量上方的名称都与 AF3 补充中使用的张量名称相匹配。通常,张量在通过模型时会保留其名称。然而,在某些情况下,我们使用不同的名称来区分张量在不同处理阶段的版本。例如,在原子级 single 表示中, c 表示初始原子级 single 表示,而 q 表示该表示在 Atom Transformer 中进行时的更新版本。
- 为了简单起见,我们还忽略了大多数 LayerNorm,但它们被使用 到处.
1. 输入准备

用户实际提供给 AF3 的输入是一条蛋白质序列,以及可选的其他分子。本节的目标,是把这些序列转换成 6 个张量,作为模型主干的输入:s,token 级 single 表示;z,token 级 pair 表示;q,原子级 single 表示;p,原子级 pair 表示;m,MSA 表示;以及 t,template 表示。
本节包含:
- Token 化 描述分子如何 Token 化并阐明原子级和 token 级之间的区别
- 检索(创建 MSA 和模板) 解释了我们为何以及如何在模型中包含额外的输入。它创建了我们的 MSA (m)和结构 template(t).
- 创建原子级表示 创建我们的第一个原子级表示 q (单个)和 p (对)并包括有关分子生成的构象的信息。
- 更新原子级表示(Atom Transformer) 是主要的 “Input Embedder” 模块,也叫 “Atom Transformer”。它会重复 3 次,并更新原子级 single 表示(q)。这里介绍的构建块(Adaptive LayerNorm、Attention with Pair Bias、Conditioned Gating 和 Conditioned Transition)在后续模型中也会用到。
- 聚合原子级 -> token 级 采用我们的原子级表示(q, p)并聚合多原子 token 一部分的所有原子以创建 token 级表示 s (单个)和 z (对)并包括来自 MSA 的信息(m)以及任何用户提供的有关涉及配体的已知键的信息。
Token 化


在 AF2 中,由于模型仅表示具有一组固定氨基酸的蛋白质,因此每个氨基酸都用自己的 token 表示。这在 AF3 中得到了维护,但还为 AF3 可以处理的其他分子类型引入了其他 token:
- 标准氨基酸:1 个 token(根据 AF2)
- 标准核苷酸:1 个 token
- 非标准氨基酸或核苷酸(甲基化核苷酸、翻译后修饰的氨基酸等):1个 token 每个原子
- 其他分子:1个 token 每个原子
因此,我们可以将某些 token(例如氨基酸的 token)视为与多个原子相关联,而其他 token(例如配体中的原子的 token)仅与单个原子相关联。因此,虽然具有 35 个标准氨基酸(可能 > 600 个原子)的蛋白质将由 35 个 token 表示,但具有 35 个原子的配体也将由 35 个 token 表示。
检索(创建 MSA 和模板)

AF3 的关键早期步骤之一类似于检索增强生成 RAG 在语言模型中。我们找到与我们感兴趣的蛋白质和 RNA 序列相似的序列(收集到多序列比对中,“MSA”),以及与这些序列相关的任何结构(称为“模板”),然后将它们作为附加输入添加到称为“模板”的模型中。 m and t,分别。

为什么我们要包含 MSA 和模板?
在不同物种中发现的相同蛋白质的版本在结构和顺序上可能非常相似。通过将它们排列在一起形成多重序列比对 (MSA),我们可以了解蛋白质序列中的单个位置在整个进化过程中如何变化。你可以将给定蛋白质的 MSA 视为矩阵,其中每一行都是来自不同物种的类似蛋白质的序列。研究表明,沿着蛋白质中特定位置的柱发现的保守模式可以反映该位置存在某些氨基酸的重要性,并且不同柱之间的关系反映了氨基酸之间的关系(即,如果两个氨基酸在物理上相互作用,则它们的氨基酸的变化可能在进化过程中相关)。因此,MSA 通常用于丰富单个蛋白质的表达。
同样,如果这些蛋白质中的任何一个具有已知的结构,这些也可能告知该蛋白质的结构。不寻找完整的结构,而是仅使用蛋白质的单个链。这类似于同源建模的实践,其中查询蛋白质的结构是基于来自假定相似的已知蛋白质结构的模板来建模的。
那么这些序列和结构是如何检索的呢?
首先,进行遗传搜索,寻找与任何输入蛋白质或 RNA 链相似的任何蛋白质或 RNA 链。这不涉及任何训练,并且依赖于现有的基于隐马尔可夫模型(HMM)的方法
与 AF 多聚体相比,这些检索步骤的唯一新部分是我们现在除了蛋白质序列之外还对 RNA 序列进行检索。请注意,这在传统上并不被称为“检索”,因为使用结构 template 来指导蛋白质结构建模的做法在以下领域已是常见做法: 同源建模 早在 RAG 一词出现之前。然而,尽管 AlphaFold 没有明确地将这个过程称为检索,但它确实非常类似于现在流行的 RAG。
我们如何表示这些模板?
从我们的模板搜索中,我们获得了每个模板的 3D 结构以及有关哪些 token 位于哪些链中的信息。首先,计算给定模板中所有 token 对之间的欧氏距离。对于与多个原子关联的 token,一个代表 “中心原子” 用于计算距离。这将是 Cɑ 氨基酸的原子和 C1’ 标准核苷酸的原子。

这会生成一个 Ntoken xNtoken 每个模板的矩阵。然而,不是将每个距离表示为数值,而是将距离离散化为“直方图”(距离直方图)。
然后,我们向每个分布图添加有关哪个链的元数据
创建原子级表示


创造 q,我们的原子级 single 表示,我们需要提取所有原子级特征。第一步是计算每个氨基酸、核苷酸和配体的“参考构象”。虽然我们还不知道整个复合体的结构,但我们对每个单独组件的局部结构有很强的先验知识。构象(简称 构象) 是分子中原子的 3D 排列,是通过对单键旋转采样生成的。每个氨基酸都有一个“标准”构象,它只是该氨基酸可以存在的低能构象之一,可以通过查找来检索。然而,每个小分子都需要自己的构象生成。这些是用生成的 RDKit 的 ETKDGv3,一种结合实验数据和扭转角度偏好来生成 3D 构象的算法。
然后,我们把该构象中的相对位置信息,与每个原子的电荷、原子序数以及其他标识符拼接起来。矩阵 c 存储序列中所有原子的这些信息。随后,我们用 c 初始化原子级 pair 表示 p,用于保存原子之间的相对距离。由于此时只知道同一 token 内部的参考距离,模型会使用掩码 v,确保这个初始距离矩阵只包含构象生成阶段实际计算过的距离。模型还会加入距离平方倒数的线性嵌入、cl 和 cm 的投影,并用带残差连接的线性层继续更新它。AF3 论文没有详细解释这个额外的反距离步骤为什么必要,也没有给出对应消融;和后面许多设计一样,我们只能推测它在经验上确实有用。
最后,模型复制一份原子级 single 表示,并把这份副本命名为 q。之后真正被更新的是 q,而初始表示 c 会被保留下来,供后续步骤继续使用。
更新原子级表示(Atom Transformer)
已生成 q (所有原子的代表)和 p (每对原子的表示),我们现在想要根据附近的其他原子更新这些表示。每当 AF3 在原子级别应用注意力时,我们都会使用一个名为 Atom Transformer 的模块。Atom Transformer 是一系列使用注意力来更新的块 q 两者同时使用 p 和原始表示 q called c。作为 c 注意力变换器不会更新,它可以被认为是与起始表示的剩余连接。
Atom Transformer 主要遵循标准 Transformer 结构,使用层规范、注意力,然后是 MLP 转换。然而,每个步骤都经过了调整,以包含来自以下方面的额外输入: c and p (这里包括辅助输入有时被称为“条件”。)注意力模块和 MLP 模块之间还有一个“门控”步骤。更详细地完成这 4 个步骤中的每一个步骤:
1. Adaptive LayerNorm


Adaptive LayerNorm (AdaNorm) 是 LayerNorm 的一种变体,具有一个简单的扩展。回想一下,对于给定的输入矩阵,传统的 LayerNorm 学习两个参数(缩放因子 gamma 和偏差因子 beta),用于调整矩阵中每个通道的平均值和标准差。 AdaNorm 不是学习 gamma 和 beta 的固定参数,而是学习一个基于输入矩阵自适应生成 gamma 和 beta 的函数。然而,不是根据重新缩放的输入生成参数(在 Atom Transformer 中,这是 q),辅助输入(c 在 Atom Transformer 中)用于预测重新调整平均值和标准差的 gamma 和 beta q.
2. Attention with Pair Bias

具有 pair bias 的原子级注意力可以被认为是自注意力的延伸。就像在自注意力中一样,查询、键和值都来自相同的一维序列(我们的 single 表示, q)。但是,有 3 个区别:
-
成对偏压:计算查询和键的点积后,添加 pair 表示的线性投影作为偏差来缩放注意力权重。请注意,此操作不涉及任何信息 q 被用来更新 p,只有一种方式从 pair 表示流到 q。这样做的原因是,具有更强成对关系的原子应该更强烈地相互关注,并且 p 实际上已经编码了注意力图。
-
Gating:除了查询、键和值之外,我们还创建了一个额外的投影 q 它通过 sigmoid 传递,将值压缩在 0 和 1 之间。我们的输出在所有头重新组合之前乘以这个“门”。这有效地迫使模型忽略它在注意力过程中学到的一些东西。这种类型的门控在 AF3 中经常出现,并且在 ML 思考部分进行了更多讨论。简单来说,由于模型不断地将每个部分的输出添加到残差流中,因此可以将这种门控机制视为模型指定在该残差流中保存或不保存哪些信息的方式。它可能以 LSTM 中类似的“门”命名为“门”,LSTM 使用 sigmoid 来学习过滤器,以了解将哪些输入添加到运行的单元状态中。
-
注意力稀疏:
| 因为原子的数量可能比 token 的数量大得多,所以我们在这一步不会运行完全注意力,而是使用一种稀疏注意力(称为序列局部原子注意力),其中注意力有效地在局部组中运行,其中一次 32 个原子的组可以全部关注其他 128 个原子。更彻底地描述稀疏注意力模式 互联网上的其他地方. |  |
3. Conditioned Gating

我们对数据应用另一个门,但这次门是从我们的原始原子级单矩阵生成的, c
4. Conditioned Transition
此步骤相当于 Transformer 中的 MLP 层,之所以称为“条件”,是因为 MLP 夹在 Adaptive LayerNorm(Atom Transformer 的步骤 1)和 Conditioned Gating(Atom Transformer 的步骤 3)之间,两者都依赖于 c.
本节中唯一需要注意的是 AF3 在转换块中使用 SwiGLU 而不是 ReLU。从 ReLU → SwiGLU 的转变发生在 AF2 → AF3 中,并且是许多最新架构中的常见变化,因此我们在这里将其可视化。
使用基于 ReLU 的过渡层(如 AF2 中),我们获取激活值,将它们投影到 4 倍大小,应用 ReLU,然后将它们向下投影回原始大小。当使用 SwiGLU(在 AF3 中)时,输入激活会创建两个中间上投影,其中一个经过 swish 非线性(ReLU 的改进变体),然后在下投影之前将它们相乘。下图显示了差异:

聚合原子级 → token 级


虽然到目前为止的数据都存储在原子级别,但 AF3 的表示学习部分从现在开始在 token 级别运行。为了创建这些 token 级表示,我们首先将原子级表示投影到更大的维度(catom=128,ctoken=384)。然后,我们取分配给同一 token 的所有原子的平均值。请注意,这仅适用于与标准氨基酸和核苷酸相关的原子(通过对附加到同一 token 的所有原子取平均值),而其余部分保持不变
现在我们已经进入 “token 空间”。模型会把 token 级特征与 MSA 中可用的统计量拼接起来

现在我们已经创建了 sinit,我们初始化的 single 表示,下一步是初始化我们的 pair 表示 zinit。pair 表示是一个三维张量,但最容易将其视为隐式深度维度为 c 的类似热图的二维矩阵z=128 个频道。那么,进入 zi,j 我们的 pair 表示是 cz 维度向量旨在存储有关 token 序列中 token i 和 token j 之间关系的信息。我们创建了一个类似的原子级矩阵 p,我们在 token 级别遵循类似的过程。
初始化 zi,j,我们使用线性投影使序列表示的通道维度与 pair 表示的通道维度 (384 → 128) 相匹配,并将结果相加 si and sj。为此,我们添加相对位置编码, pi,j
现在我们已经成功创建并嵌入了将在模型的其余部分中使用的所有输入:

对于第 2 步,我们将保留原子级表示(c, q, p)并专注于更新我们的 token 级表示 s and z 在下一节中(在 m and t).
2.表征学习

此部分是模型的主要部分,通常称为“主干”,因为大部分计算都是在此处完成的。我们将其称为模型的表示学习部分,因为目标是学习 token 级“单个”的改进表示(s)和“对”(z) 上面初始化的张量。
本节包含:
- Template 模块 updates z 使用结构 template t
- MSA 模块 首先更新 MSA m,然后将其添加到 token 级 pair 表示中 z。在本节中,我们在两个操作上花费了大量时间:
- 外积均值 enables m 影响 z
- 仅使用 pair bias 的 MSA 行式门控自注意力 updates m 基于 z 是具有 pair bias 的注意力的简化版本(用于 MSA)
- Pairformer updates s and z 具有几何启发(三角形)的注意力。本节主要描述三角形运算(在 AF2 和 AF3 中广泛使用)。
- 为什么要看三角形? 解释了三角形运算的一些直觉
- Triangle Updates and Triangle Attention 都更新 z 使用类似于自注意力的方法,但受到三角不等式的启发
- Single Attention with Pair Bias updates s 基于 z 并且是具有 pair bias 的注意力的 token 级等价物(用于单个序列)
每个单独的块都会重复多次,然后整个部分的输出再次作为输入反馈到自身,并重复该过程(这称为 Recycling)。
Template 模块


每个 template(图中为 N_template=2)都会先经过一次线性投影,并与 pair 表示(z)的线性投影相加。这个新组合出的矩阵会经过一系列称为 Pairformer Stack 的操作(后文会详细介绍)。最后,所有 template 的结果会先取平均,再通过另一个线性层。
MSA 模块


该模块非常类似于 AF2 中的“Evoformer”,其目标是同时改进 MSA 和 pair 表示。它对这两种表示独立地执行一系列操作,然后还可以实现它们之间的串扰。
第一步是对 MSA 的行进行二次采样,而不是使用之前生成的 MSA 的所有行(最多可达 16k),然后将 single 表示的投影版本添加到该二次采样的 MSA 中。
Outer Product Mean

接下来,模型通过 “Outer Product Mean” 把 MSA 表示并入 pair 表示。比较 MSA 中的两列,可以揭示序列中两个位置之间的关系(例如它们在进化序列中是否相关)。对于每一对 token 索引 i,j,模型遍历所有进化序列,计算 ms,i 与 ms,j 的外积,然后在所有进化序列上取平均。之后,模型会把这个外积结果展平、投影回所需维度,并加到 pair 表示 zi,j 上。单个外积只比较同一条序列 ms 内的值;但对所有序列取平均之后,跨序列的信息就被混合进来了。
Row-wise Gated Self-Attention using only Pair Bias

根据 MSA 更新 pair 表示后,模型接下来根据 pair 表示更新 MSA。这种特定的更新模式称为 行式门控自注意力 仅使用 pair bias,并且是一个简化版本 自我关注 具有 pair bias,在 Atom Transformer 部分中讨论,独立应用于 MSA 中的每个序列(行)。它受到注意力的启发,但我们不使用查询和键来确定每个 token 应该关注的其他位置,而是使用存储在 pair 表示中的 token 之间的现有关系 z.

在 pair 表示中,每个 zi,j 是一个向量,包含有关 token i 和 j 之间关系的信息。当张量 z 被投影到一个矩阵,每个 zi,j 向量成为一个标量,可用于确定 i 应该关注 token j 的程度。应用 row-wise softmax 后,这些现在相当于注意力分数,用于创建值的加权平均值,就像典型的注意力图一样。
请注意,MSA 中的进化序列之间没有共享信息,因为它是针对每一行独立运行的。
更新 pair 表示
MSA 模块的最后一步是通过一系列称为Triangle Updates 和 Triangle Attention的步骤来更新 pair 表示。下面通过 Pairformer 描述这些三角形运算,并再次使用它们。还有一些转换块使用 SwiGLU 向上/向下投影矩阵,就像在 Atom Transformer 中所做的那样。
Pairformer 模块


AF3 补充材料图
使用 template 和 MSA 更新 pair 表示后,模型后续就不再直接使用它们。进入 Pairformer 的只有更新后的 pair 表示(z)和 single 表示(s),二者会在 Pairformer 中相互更新。由于 transition block 前面已经介绍过,本节重点放在 Triangle Updates 和 Triangle Attention 上,然后简要说明 Single Attention with Pair Bias 与前文版本的区别。这些基于三角关系的层最早出现在 AF2 中,不仅在 AF3 中保留下来,还变得更加核心,因此值得重点展开。
为什么要看三角形?
这里的核心直觉来自三角不等式:“三角形任意两边之和大于或等于第三边”。回忆一下,每个 zi,j 都编码了序列中位置 i 和 j 之间的关系。它并不字面表示两个 token 的物理距离,但我们可以先用距离来类比:如果 zi,j=1 且 zj,k=1,那么按三角不等式,zi,k 不可能大于 2。知道两条边的信息,会强烈约束第三条边。Triangle Updates 和 Triangle Attention 的作用,就是让模型能利用这种三元关系带来的几何约束。
模型并不会硬性强制满足三角不等式,而是通过信息流鼓励这种一致性:每个位置 zi,j 都会参考所有可能的三元组 (i,j,k) 来更新。因此,zi,j 可以利用所有其他 token k 对应的 zj,k 和 zi,k 信息。由于 z 表示的不只是距离,而是 token 之间复杂且可能有方向的物理关系,所以模型也会从 zk,i 和 zk,j 中收集信息。如果把 token 看成图中的节点,把 z 看成有向邻接矩阵,那么 AlphaFold 把这些操作称为 “outgoing edges” 和 “incoming edges” 就很自然。
考虑该邻接矩阵的第 i=0 行,假设我们要更新 z0,2,已以紫色突出显示。更新背后的想法是,如果我们知道 0→1 和 2→1 之间的距离,就会给我们一些关于 0→2 的限制。类似地,如果我们知道 0→3 和 2→3 之间的距离,这也给了我们对 0→2 的约束。这适用于所有token k。

因此,在Triangle Updates 和 Triangle Attention中,我们有效地查看该图中 3 个节点的所有有向路径(也就是三角形,名称也由此而来。)。

Triangle Updates
从图论的角度仔细研究了三角形运算,我们可以看到这是如何通过张量运算实现的。在即将发布的更新中,每个位置 zi,j 在 pair 表示中,根据同一行中其他元素的加权组合独立更新(zi,j),其中每个的权重 zi,k 基于其出边三角形中的第三个元素(zj,k).

实际上,我们采用三个线性投影 z (称为 a、b 和 g)。更新 zi,j,我们进行逐元素乘法 第 i 行,来自 a and b 行 j。然后我们对所有这些行(不同的 k 值)求和,并使用 g 投影进行门控。

对于传入的更新,我们有效地执行相同的操作,但将行与列翻转,以便更新 zi,j 我们对同一列中的其他元素进行加权和(zk,j),其中每个的权重 zk,j 基于其出边三角形中的第三个元素(zk,i)。创建相同的线性投影后,我们进行逐元素乘法 column 我从 a 和 column 来自 b 的 j,并对所有的求和 这个矩阵的行。你会发现这些操作完全反映了上述图论邻接视图。
Triangle Attention
在我们的两个三角形更新步骤之后,我们还更新每个 zi,j using 三角注意力 用于传出边缘,Triangle Attention 用于传入边缘。 AF3 论文将“传出边缘”称为“围绕起始节点”的注意力,将“传入边缘”称为“围绕结束节点”的注意力。

为了建立 Triangle Attention,从一维序列上的典型自注意力开始可能会有所帮助。回想一下,查询、键和值都是原始一维序列的转换。一种名为“注意力”的变体 轴向注意力 通过在 2D 矩阵的不同轴(行,然后列)上应用独立的 1D 自注意力,将其扩展到矩阵。Triangle Attention在此基础上添加了我们之前讨论的三角形原理,更新 zi,j 通过合并 zi,k and zj,k 对于所有token k。具体来说,在“起始节点”的情况下,计算沿第 i 行的注意力分数(以确定有多少 zi,j 应该受到影响 zi,k),我们进行查询键比较 zi,j and zi,k 像往常一样,然后根据 zj,k 如上图所示。

对于“结束节点”的情况,我们再次将行交换为列。对于 zi,j,键和值都来自第 i 列 z,而偏差将来自第 j 列。因此,在比较查询时 zi,j 用钥匙 zk,i,我们根据注意力分数来偏置 zk,j。然后,一旦我们获得了所有 k 的注意力分数,我们就使用第 i 列中的值向量。
Single Attention with Pair Bias

现在我们已经用这四个三角形步骤更新了 pair 表示,我们将 pair 表示通过 Transition 块传递,如上所述。最后,我们想要更新我们的 single 表示(s)使用这个新的更新的 pair 表示(z),所以我们将使用带有 pair bias 的 Single Attention,如下图所示。这与描述的 Single Attention with Pair Bias 相同
我们重复 48 个区块的配对,最终创建 strunk and ztrunk.
3. 结构预测
扩散基础
现在,有了这些经过充分更新的表示,模型就可以使用 s 和 z 来预测复合物结构。AF3 的一个重要变化是:整个结构预测都基于原子级扩散。已有文章更系统地解释了扩散模型的直觉和数学细节;这里只保留核心思想:从真实数据出发,逐步加入随机噪声,并训练模型预测加入了哪些噪声。噪声会在一系列 T 个时间步中逐步加入数据,形成每个数据点的 T 个带噪版本。原始数据记作 x0,完全加噪的版本记作 xt=T。训练时,在时间步 t,模型看到 xt,并预测从 xt-1 到 xt 之间加入了多少噪声;然后用预测噪声和真实加入的噪声之间的差异来更新模型。
然后,在推理时,我们简单地从随机噪声开始,这相当于 xt=T。对于每个时间步,我们预测模型认为已添加的噪声,并删除预测的噪声。经过预先指定的时间步数后,我们最终得到一个完全“去噪”的数据点,它应该类似于数据集中的原始数据。
条件扩散让模型根据某些输入“条件”这些去噪预测。实际上,这意味着对于模型的每个步骤,它需要三个输入:
- 我们这一代当前嘈杂的迭代
- 我们当前所处时间步的表示
- 我们想要调节的信息(这可能是要生成的图像的标题,或者蛋白质的属性)。
因此,最终的生成不仅仅是一个类似于训练数据分布的随机示例,而且应该专门匹配该条件向量表示的信息。
使用 AF3,我们学习去噪的数据是一个矩阵 x 序列中所有原子的 x、y、z 坐标。在训练过程中,我们向这些坐标添加高斯噪声,直到它们实际上完全随机。然后在推理时,我们从随机坐标开始。在每个时间步,我们首先随机旋转并平移整个预测的复合体。这种数据增强告诉模型,我们的复合体的任何旋转和平移都同样有效,并取代了 AF2 中使用的更复杂的不变点注意力。

扩散模块


在每个去噪扩散步骤中,我们根据输入序列的多种表示来调整预测:
- 主干的输出(我们的后配对器更新 s and z,现在称为 strunk and ztrunk)
- 在输入嵌入器中创建的序列的初始原子和 token 级表示尚未通过主干(sinputs, cinputs)
AF3 论文将其扩散过程分为 4 个步骤,涉及从 token 到原子、回到 token、再回到原子:
- 准备 token 级调节张量
- 准备原子级条件张量,使用 Atom Transformer 更新它们,并将它们聚合回 token 级
- 在 token 级别应用注意力,然后投射回原子
- 在原子级应用注意力来预测原子级噪声更新
1. 准备 token 级调节张量


为了初始化我们的 token 级条件表示,我们连接 ztrunk 到相对位置编码,然后将这个更大的表示投影回并通过几个残差连接转换块。
类似地,对于我们的 token 级 single 表示,我们连接在模型开始时创建的输入的第一个表示(sinputs)和我们目前的代表(strunk),然后将其投影回原来的大小。然后,我们根据当前扩散时间步创建傅里叶嵌入
2. 准备原子级张量,应用原子级注意力,并聚合回 token 级
此时,我们的条件向量在每个 token 级别存储信息,但我们也希望在原子级别运行注意力。为了解决这个问题,我们采用嵌入部分中创建的输入的初始原子级表示(c and p),并根据当前的 token 级表示更新它们,以创建原子级条件张量。


接下来,我们缩放原子的当前坐标(x)通过数据的方差,有效地创建具有单位方差的“无量纲”坐标(称为 r)。然后我们更新 q 基于 r 这样 q 现在知道原子的当前位置。最后我们更新一下 q 使用 Atom Transformer(它也将 pair 表示作为输入),并将原子聚合回 token,如我们之前所见。

在这一步结束时,我们返回
- q:合并有关原子当前坐标的信息后更新了原子表示
- a:token 级别的聚合形式 q,捕获坐标和序列信息
- c:基于主干的调节的原子表示
- p:我们更新的用于调节的原子 pair 表示
3. 在 token 级应用注意力
此步骤的目标是应用注意力来更新原子坐标和序列信息的 token 级表示, a。此步骤使用在输入准备期间可视化的扩散 Transformer,它镜像原子 Transformer,但用于 token。
4. 在原子级应用注意力来预测原子级噪声更新
现在回到原子空间。模型使用更新后的 a(基于当前“中心原子”位置得到的 token 级表示)来更新 q(基于当前位置的原子级表示)。和步骤 3 一样,模型会把 token 表示广播到原子数维度:如果一个 token 表示多个原子,就按需复制它的表示,然后运行 Atom Transformer。最后,线性层把这个原子级表示 q 映射回 R3。这是关键步骤:模型利用所有条件表示,为所有原子生成坐标更新 rupdate。现在就可以把这些更新加回当前带噪坐标,进入下一个去噪步骤。
至此,我们就完成了 AlphaFold 3 主要架构的参观!现在我们提供一些有关损失函数、辅助置信头和训练细节的附加信息。
4. 损失函数和其他训练细节
损失函数和置信度头
Lloss = Ldistogram * αdistogram + Ldiffusion * αdiffusion + Lconfidence * αconfidence
损失是三项的加权和:
- Ldistogram 它评估 token 级别的预测分布图的准确性
- Ldiffusion 它评估原子水平上预测的直方图的准确性。它着眼于所有成对距离,然后包含附加项来优先考虑附近原子和参与蛋白质-配体键的原子之间的距离。
- Lconfidence 它评估模型对哪些结构可能不准确的自我意识
L_distogram
我们模型的输出是原子级坐标,可以轻松地用于创建原子级直方图
L_diffusion
扩散损失本身是三项的加权和,每项在原子位置上计算,另外还按噪声量进行缩放
Ldiffusion = (LMSE + Lbond * αbond) * (t̂² + σdata²)/(t̂+σdata)² + Lsmooth_lddt
- LMSE 是我们刚刚讨论的直方图损失的一个版本,但对于所有原子而言,而不仅仅是“中心原子”(并且 DNA、RNA 和配体原子的权重增加)。此外,它还会查看位置之间的均方误差,而不是将它们合并到直方图中。
- Lbond 旨在通过对作为蛋白质-配体键一部分的原子对的预测和真实分布图的差异添加额外的 MSE 损失来确保蛋白质-配体键键长的准确性。
有不同的训练阶段和αbond 在初始阶段被设置为0,所以这个术语是在后面才引入的。 - Lsmooth_LDDT (平滑局部距离差异测试)是直方图损失的另一种变体,试图捕获局部距离的准确性。如果原子对的预测距离在原子对真实距离的给定阈值内,则该距离“通过测试”。为了使这个指标平滑且可微分,我们通过以测试阈值为中心的 sigmoid 函数传递预测直方图和真实直方图之间的差异。我们可以将其视为生成该原子对通过测试的概率(0 到 1 之间)。我们取四次“测试”的平均值,阈值越来越紧(4、2、1 和 0.5 Å)。使用这种损失可以鼓励模型降低每次测试失败的概率。最后,为了使测试“局部”,如果原子对的真实距离很大,我们会忽略该原子对的损失,因为我们只希望模型专注于准确预测原子到附近原子的距离
具体来说,对于原子对 l,m,如果原子 m 是核苷酸的一部分,且 l 和 m 相距超过 30 Å,则我们忽略 l 和 m 的损失。如果 l 和 m 彼此相距超过 15 Å 并且 m 不是核苷酸(因此是蛋白质或配体的一部分),我们会忽略 l 和 m 的损失。 .

L_confidence
这种损失的目标不是提高结构的准确性,而是教会模型预测其自身的准确性。该损失是 4 项的加权和,每项对应于评估预测结构质量的方法:
Lconfidence = LpLDDT + LPDE + Lresolved + LPAE * αPAE
-
lDDT 原子级“局部距离差异测试”,捕获原子与附近原子的预测距离的预期准确性。
-
PAE token i 的预测位置和真实位置之间的预测对齐误差。我们首先将预测 token i 和真实 token i 旋转并转换为 token j 的框架。也就是说,如果我们暂时假设 token j 正好处于其真实位置,我们就会根据 token i 与 token j 的关系来预测 token i 与它应在的位置有多接近。
-
PDE token 之间的预测距离误差,捕获所有 token 对之间的预测差异的准确性。
-
实验解决的预测 该模型预测哪些原子是通过实验解析的(并非每个晶体结构中的每个原子都通过实验解析)。
为了获得每个指标的置信度损失,AF3 预测这些误差指标的值,然后根据预测的结构计算这些误差指标,并且损失基于这两者之间的差异。因此,即使结构确实不正确且 PAE 高,如果预测的 PAE 也高,则 Lpae 会很低。

这些置信度预测是在扩散过程中生成的。在选定的扩散步骤 t 处,预测坐标 rt 用于更新在表示学习主干中创建的 single 表示和 pair 表示。然后根据更新的 pair 表示(PAE 和 PDE)或更新的 single 表示(pLDDT 和实验解析)的线性投影来计算预测误差。然后,基于相同生成的原子坐标计算实际误差度量(如果感兴趣,下面描述过程)以进行比较。
虽然这些项包含在置信度头损失中,但这些项的梯度仅用于更新置信度预测头,不会影响模型的其余部分。
实际的误差指标是如何计算的?
LDDT: 原子 l 的 LDDT 计算方式如下:在当前预测结构中,计算原子 l 与一组由 m 索引的原子 R 之间的距离,并与真实结构中的对应距离比较。要进入集合 R,原子 m 必须属于一条聚合物链,且距离 l 不超过 15 Å 或 30 Å(阈值取决于 m 所属分子),并且是某个 token 的中心原子。随后,模型用 4、2、1、0.5 Å 这四个逐渐严格的阈值做二元距离测试,取平均通过率,并在 R 中所有原子上求和。最后,这个百分比会被划分到 0 到 1 之间的 50 个 bin 中。
在推理时,我们有一个 pLDDT 头。该头采用给定 token 的 single 表示,在“附加”到该 token 的所有原子上重复它
预测对齐误差 (PAE): 每个 token 都被认为有一个框架,即由该 token 涉及的三个原子(称为 a、b、c)创建的 3D 坐标框架。这三个原子中的原子 b 构成了该框架中的原点。在每个 token“附加”单个原子的情况下,框架的中心原子是 token 的单个原子,并且同一实体(例如,相同配体)的其他两个最近的 token 形成框架的基础。对于每个 token 对 (i,j),我们使用 token_j 的框架重新表达 token_i 的中心原子的预测坐标。我们对 token_i 中心原子的真实坐标执行相同的操作。 token_i 中心原子的这些变换后的真实坐标和预测坐标之间的欧氏距离是我们的对齐误差,分为 64 个 bin。我们从 pair 表示中预测这种对齐误差 zi,j,将其投影到我们将其视为 logits 的 64 维,并使用 softmax 转换为概率。我们用分类损失来训练这个头,其中每个 bin 作为一个类。参见 这里 了解更多详情。
第三,AF3 预测 token 之间的距离误差(PDE)。真实距离误差的计算方法是:对每一对 token 的中心原子计算距离,并把这些距离划分到从 0 Å 到 32 Å 的 64 个等宽 bin 中。预测的距离误差来自 pair 表示 zi,j 与 zj,i 之和;模型把它投影到 64 维,作为 logits,再用 softmax 转成概率。
最后,AF3 预测每个原子是否在真实结构中通过实验得到解析。与 pLDDT 头类似,我们重复 si 该 token 代表的原子数量的 single 表示,并投影到二维并使用二元分类损失。
其他训练细节
现在已经介绍了架构,最后一部分是一些额外的训练细节。
Recycling
正如 AF2 中所介绍的,AF3 会 Recycling 其权重;也就是说,不是使模型更深,而是重新使用模型权重,并且多次通过模块运行输入以不断改进表示。扩散本质上在推理时使用 Recycling,因为模型经过训练以合并时间步信息并为每个时间步使用相同的模型权重。
交叉蒸馏
AF3 的训练数据混合了由自身生成的合成数据(self-distillation),也混合了 AF2 生成的数据(cross-distillation)。具体来说,作者指出,当结构模块切换为基于扩散的生成模块后,模型不再生成 AF2 中那种典型的“意大利面条”状区域;而这些区域原本能让 AF2 用户一眼识别低置信度、可能无序的片段。只看基于扩散的生成结果时,所有区域看起来都同样自信,这让潜在 hallucination 更难识别。
为了解决这个问题,作者把 AF2 和 AF-Multimer 的生成结果加入 AF3 训练数据,让模型学到:当 AF2 对某段结构置信度低时,它会输出这些展开区域,AF3 也应该以类似方式表达低置信度。

裁剪和训练阶段
虽然模型的任何部分对输入序列的长度没有明确的限制,但内存和计算需求随着序列长度的增加而显着增加(回想一下多个 O(Ntokens3 操作))。因此,为了提高效率,蛋白质被随机裁剪。正如 AF-Multimer 中所介绍的,因为我们想要对多个链之间的相互作用进行建模,所以随机裁剪需要包括所有这些。他们使用 3 种裁剪方法,所有 3 种方法根据训练数据以不同比例使用(例如:PDB 晶体结构与无序 PDB 复合体与蒸馏等)
- 连续裁剪:为每条链选择连续的氨基酸序列
- 空间裁剪:根据与参考原子的距离选择氨基酸(通常该原子是感兴趣的特定链或结合界面的一部分)
- 空间界面裁剪:与空间裁剪类似,但基于到特定于结合界面的原子的距离。
虽然在 384 个随机裁剪上训练的模型可以应用于更长的序列,但为了提高模型处理这些序列的能力,它会针对更大的序列长度进行迭代微调。每个训练阶段的数据集和其他训练细节的组合也各不相同,如下表所示。

原子冲突
作者指出,AF3 的损失不包括重叠原子的碰撞惩罚。虽然切换到基于扩散的结构模块意味着该模型理论上可以预测两个原子位于同一位置,但在训练后这似乎是最小的。也就是说,AF3 在对生成的结构进行排名时确实采用了冲突惩罚。
批量大小
尽管扩散过程听起来相当复杂,但它的计算成本仍然比模型主干要低得多。因此,AF3 作者发现,从训练的角度来看,在主干之后扩大模型的批量大小会更有效。因此,对于每个输入结构,它都会通过嵌入和主干运行,然后应用该结构的 48 个独立的数据增强版本,并且这 48 个结构都是并行训练的。
这就是训练过程! 还有一些其他的小细节,但这可能已经超出了你的需要,如果你已经做到了这一点,那么其余的内容应该很容易通过阅读 AF3 补充材料来掌握。
机器学习思考
在彻底了解 AF3 的架构及其与 AF2 的比较之后,作者所做的选择如何适应更广泛的机器学习趋势,这一点很有趣。
AlphaFold 作为检索增强生成
AF2 发布时,在推理时从训练集中进行检索并不常见。对于 AF,利用 MSA 和模板搜索。基于 MSA 的方法被用于蛋白质建模,但这种类型的检索在深度学习的其他领域中较少使用(例如,在计算机视觉中对新图像进行分类时,ResNet 在推理时不会嵌入相关的训练图像)。尽管与 AF2 相比,AF3 减少了对 MSA 的重视(不再在 Evoformer/Pairformer 的 48 个模块中进行操作和更新),但它们仍然包含 MSA 和模板,即使其他蛋白质预测模型(例如 ESMFold)已经放弃检索,转而支持全参数推理。
有趣的是,一些最大、最成功的深度学习模型现在通常在推理时包含类似的附加信息。虽然检索系统的细节并不总是公开,但大型语言模型通常在推理时使用检索增强生成(RAG)系统(例如传统的网络搜索)来将模型定位到应该指导推理的相关信息(即使该信息可能已经在其训练数据中)。看看未来如何在推理时使用直接相关的示例将会很有趣。
Pair-Bias Attention
AF2 的主要组成部分之一(在 AF3 中更常见)是 Pair-Bias Attention(Pair-Bias Attention)。也就是说,注意力的查询、键和值都源自同一来源(如自注意力),但有一个偏差项从另一个来源添加到注意力图中。这有效地充当了信息共享的轻触版本,无需完全交叉关注。 Pair-Bias Attention 几乎出现在每个模块中。虽然这种类型的注意力现在已用于其他蛋白质建模架构中,但我们还没有看到这种特定类型的交叉偏差在其他领域中使用(尽管这并不意味着它还没有完成!)。也许它只在这里工作得很好,因为 pair 表示自然地类似于自注意力图,但它是纯自注意力或纯交叉注意力的有趣替代品。
自监督训练
通过使用自监督预训练将 MSA 嵌入替换为“概率 MSA”,像 ESM 这样的自监督模型已经能够在预测蛋白质结构方面取得令人印象深刻的结果。在 AF2 中,模型有一项额外的任务,即预测来自 MSA 的屏蔽 token,实现类似的自我监督,但在 AF3 中被删除了。我们还没有看到作者评论为什么他们没有在 MSA 上使用任何自监督语言建模预训练方法,并且实际上减少了用于处理 MSA 的计算量。不使用自监督学习来初始化 MSA 嵌入的三个可能原因是 1) 他们认为大规模预训练阶段是对计算的次优使用 2) 他们进行了尝试,发现包含一个小型 MSA 模块优于预训练的嵌入,并且值得额外的推理时间成本或 3) 混合使用氨基酸 token 的预训练嵌入和随机初始化的嵌入 DNA/RNA/配体在其混合原子 token 结构上的完全监督训练中不兼容或表现不佳。
分类与回归
与 AF2 一样,AF3 继续混合使用 MSE 和分级分类损失。分类损失部分很有趣,因为如果模型预测的 distogram bin 仅相差一,则它不会因为“只差一点”而得到额外奖励。目前还不清楚这个设计决策的依据是什么,但也许作者发现梯度比使用几种不同的 MSE 损失更稳定,而且也许每个原子损失看到了如此多的梯度步骤,以至于来自连续损失的额外信号不会被证明是有益的。
与循环架构(例如 LSTM)的相似之处
AF3 的架构融合了几个设计元素,让人想起传统 Transformer 中通常不存在的循环神经网络:
- 广泛的门控:AF3 在其整个架构中使用门控机制来控制残余流中的信息流。这更类似于 LSTM 或 GRU 中的门控,而不是普通 Transformer 层的标准前馈特性。
- 具有权重重用的迭代处理:AF3 多次应用相同的权重以逐步完善其预测。这个过程涉及 Recycling 和扩散模型,类似于循环网络如何使用一组共享权重处理时间步的顺序数据。它与标准 Transformer 不同,标准 Transformer 通常在单个前向传递中进行预测。这种方法允许 AF3 在不增加参数数量的情况下迭代改进其蛋白质结构预测。
- 自适应计算:Recycling 也类似于扩散中使用的迭代更新,与自适应计算时间的思想非常相关 (ACT),最初引入是为了动态确定 RNN 使用多少计算量,最近用于 Mixture-of-Depths 用 Transformer 实现类似的目标。这与标准 Transformer 的固定深度形成鲜明对比,理论上将允许模型对具有挑战性的输入应用更多处理。
AF2 消融表明 Recycling 很重要,但很少讨论门控的重要性。据推测,它有助于提高 LSTM 中的训练稳定性,但有趣的是,它在这里如此普遍,但在许多其他基于 Transformer 的架构中却没有。
交叉蒸馏
用 AF2 的生成结果,专门把它在低置信度区域的独特表现重新引入 AF3,这一点很有意思。如果这里有什么经验教训,那可能也是最实用的一条:如果旧模型在某个具体方面比新模型做得更好,可以尝试用 cross-distillation 让新模型继承这部分优点。